NLMEResults
Results object containing estimation results from nonlinear mixed-effects modeling
Description
The NLMEResults object contains estimation results from fitting
a nonlinear mixed-effects model using sbiofitmixed.
Creation
Use the sbiofitmixed function to create an
NLMEResults object.
Properties
Object Functions
boxplot | Create box plot showing the variation of estimated SimBiology model parameters |
covariateModel | Return a copy of the covariate model that was used for the nonlinear mixed-effects
estimation using sbiofitmixed |
fitted | Return the simulation results of a fitted nonlinear mixed-effects model |
plot | Compare simulation results to the training data, creating a time-course subplot for each group |
plotActualVersusPredicted | Compare predictions to actual data, creating a subplot for each response |
plotParameterStats | Show box plot, violin plot, and swarm scatter plots for parameter estimates |
plotResidualDistribution | Plot the distribution of the residuals |
plotResiduals | Plot the residuals for each response, using the time, group, or prediction as the x-axis |
predict | Simulate and evaluate fitted SimBiology model |
random | Simulate a SimBiology model, adding variations by sampling the error model |
summary | Return parameter estimates and fit quality statistics from SimBiology nonlinear mixed-effects estimation |
Examples
This example uses data collected on 59 preterm infants given phenobarbital during the first 16 days after birth [1]. Each infant received an initial dose followed by one or more sustaining doses by intravenous bolus administration. A total of between 1 and 6 concentration measurements were obtained from each infant at times other than dose times, for a total of 155 measurements. Infant weights and APGAR scores (a measure of newborn health) were also recorded.
Load the data.
load pheno.mat ds
Convert the dataset to a groupedData object, a container for holding tabular data that is divided into groups. It can automatically identify commonly used variable names as the grouping variable or independent (time) variable. Display the properties of the data and confirm that GroupVariableName and IndependentVariableName are correctly identified as 'ID' and 'TIME', respectively.
data = groupedData(ds); data.Properties
ans = struct with fields:
Description: ''
UserData: []
DimensionNames: {'Observations' 'Variables'}
VariableNames: {'ID' 'TIME' 'DOSE' 'WEIGHT' 'APGAR' 'CONC'}
VariableTypes: ["double" "double" "double" "double" "double" "double"]
VariableDescriptions: {}
VariableUnits: {}
VariableContinuity: []
RowNames: {}
CustomProperties: [1×1 matlab.tabular.CustomProperties]
GroupVariableName: 'ID'
IndependentVariableName: 'TIME'
Create a simple one-compartment PK model with bolus dosing and linear clearance to fit such data. Use the PKModelDesign object to construct the model. Each compartment is defined by a name, dosing type, a clearance type, and whether or not the dosing requires a lag parameter. After constructing the model, you can also get a PKModelMap object map that lists the names of species and parameters in the model that are most relevant for fitting.
pkmd = PKModelDesign; addCompartment(pkmd,'Central','DosingType','Bolus',... 'EliminationType','linear-clearance',... 'HasResponseVariable',true,'HasLag',false); [onecomp, map] = pkmd.construct;
Describe the experimentally measured response by mapping the appropriate model component to the response variable. In other words, indicate which species in the model corresponds to which response variable in the data. The PKModelMap property Observed indicates that the relevant species in the model is Drug_Central, which represents the drug concentration in the system. The relevant data variable is CONC, which you visualized previously.
map.Observed
ans = 1×1 cell array
{'Drug_Central'}
Map the Drug_Central species to the CONC variable.
responseMap = 'Drug_Central = CONC';The parameters to estimate in this model are the volume of the central compartment Central and the clearance rate Cl_Central. The PKModelMap property Estimated lists these relevant parameters. The underlying algorithm of sbiofitmixed assumes parameters are normally distributed, but this assumption may not be true for biological parameters that are constrained to be positive, such as volume and clearance. Specify a log transform for the estimated parameters so that the transformed parameters follow a normal distribution. Use an estimatedInfo object to define such transforms and initial values (optional).
map.Estimated
ans = 2×1 cell
{'Central' }
{'Cl_Central'}
Define such estimated parameters, appropriate transformations, and initial values.
estimatedParams = estimatedInfo({'log(Central)','log(Cl_Central)'},'InitialValue',[1 1]);Each infant received a different schedule of dosing. The amount of drug is listed in the data variable DOSE. To specify this dosing during fitting, create dose objects from the data. These objects use the property TargetName to specify which species in the model receives the dose. In this example, the target species is Drug_Central, as listed by the PKModelMap property Dosed.
map.Dosed
ans = 1×1 cell array
{'Drug_Central'}
Create a sample dose with this target name and then use the createDoses method of groupedData object data to generate doses for each infant based on the dosing data DOSE.
sampleDose = sbiodose('sample','TargetName','Drug_Central'); doses = createDoses(data,'DOSE','',sampleDose);
Fit the model.
[nlmeResults,simI,simP] = sbiofitmixed(onecomp,data,responseMap,estimatedParams,doses,'nlmefit');Visualize the fitted results using individual-specific parameter estimates.
plot(nlmeResults,'ParameterType','individual');

Visualize the fitted results using population parameter estimates.
plot(nlmeResults,'ParameterType','population');
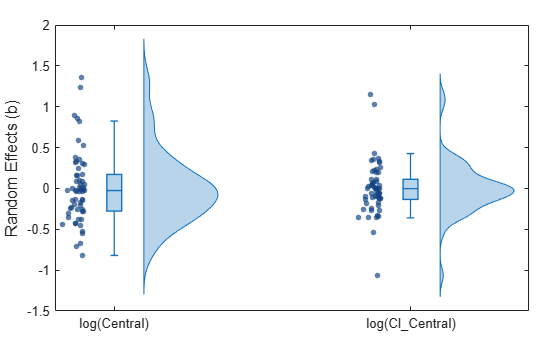
Display the statistics of parameter estimates using box plots, swarm scatter plots, and violin plots.
plotParameterStats(nlmeResults,"box","violin","swarm");
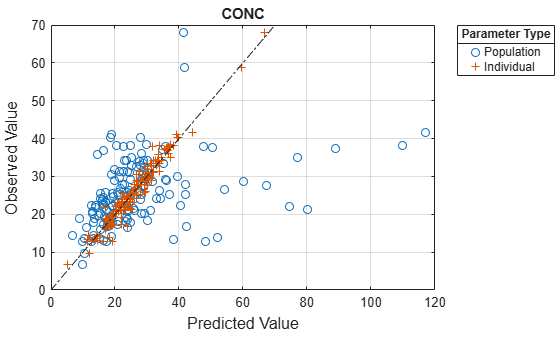
Compare the model predictions to the actual data.
plotActualVersusPredicted(nlmeResults);
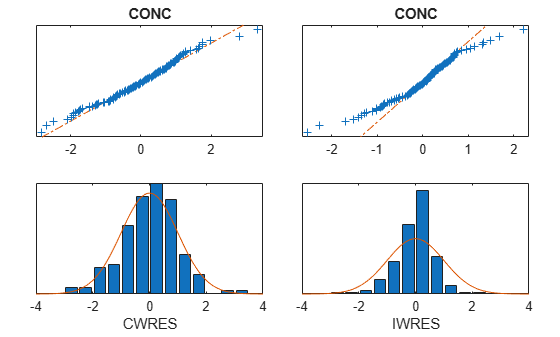
Plot the distribution of residuals.
plotResidualDistribution(nlmeResults);
Plot residuals for each response using the model predictions on x-axis.
plotResiduals(nlmeResults,'Predictions');
Get the summary of the fit results.
stats.Name contains the name for each table from stats.Table, which contains a list of tables with estimated parameter values and fit quality statistics.
fitMetrics = summary(nlmeResults);
Display the table that contains individual estimates summary statistics.
tableToShow = "Individual Estimates Summary Statistics";
tableNames = {fitMetrics.Name};
tfShowTable = matches(tableNames,tableToShow);
disp(fitMetrics(tfShowTable).Table); Statistic Central Cl_Central
______________________ _______ __________
{'Minimum' } 0.62029 0.0021127
{'Lower Quartile' } 1.0676 0.005365
{'Median' } 1.3725 0.0061236
{'Upper Quartile' } 1.6717 0.0068616
{'Maximum' } 5.4555 0.019276
{'Mean' } 1.5713 0.0064928
{'Standard Deviation'} 0.90279 0.0026232
More About
References
[1] Grasela T.H. Jr, and Donn S.M. "Neonatal population pharmacokinetics of phenobarbital derived from routine clinical data." Developmental Pharmacology and Therapeutics 8, no. 6 (1985): 374–83. https://doi.org/10.1159/000457062.
[2] Mould, D.R., and Upton, R.N. “Basic Concepts in Population Modeling, Simulation, and Model‐Based Drug Development—Part 2: Introduction to Pharmacokinetic Modeling Methods.” CPT: Pharmacometrics & Systems Pharmacology 2, no. 4 (2013): 1–14. https://doi.org/10.1038/psp.2013.14.
[3] Savic, Radojka M., and Mats O. Karlsson. “Importance of Shrinkage in Empirical Bayes Estimates for Diagnostics: Problems and Solutions.” The AAPS Journal 11, no. 3 (2009): 558–69.https://doi.org/10.1208/s12248-009-9133-0.
Version History
Introduced in R2014aSee Also
sbiofitmixed | sbiofit | nlmefit (Statistics and Machine Learning Toolbox) | nlmefitsa (Statistics and Machine Learning Toolbox)