weights
Calculate weights for phylogenetic tree
Syntax
Description
W = weights(Tree)Tree using the
Thompson-Higgins-Gibson method. The distance of every segment of the tree is adjusted by
dividing it by the number of leaves it contains. The sequence weights are the result of
normalizing to unity the new patristic distances between every leaf and the root.
Examples
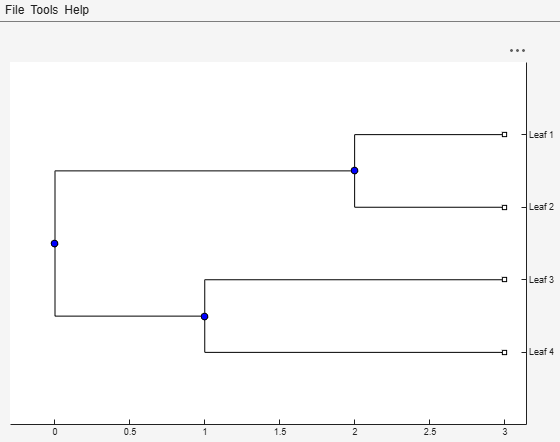
Create an ultrametric tree with specified branch distances.
bd = [1 2 3]'; tr_1 = phytree([1 2;3 4;5 6],bd)
Phylogenetic tree object with 4 leaves (3 branches)
View the tree.
view(tr_1)
Display the calculated weights.
weights(tr_1)
ans = 4×1
0.8000
0.8000
1.0000
1.0000
Input Arguments
Output Arguments
References
[1] Thompson J.D., D.G. Higgins, and T.J. Gibson. "CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice." Nucleic Acids Research 22, no. 22 (1994): 4673-4680.
[2] Henikoff S., and J.G. Henikoff (1994). "Position-based sequence weights". Journal Molecular Biology 243, no. 4 (1994): 574-578.
Version History
Introduced in R2006a
See Also
multialign | phytree | profalign | seqlinkage