Cybergenetics TrueAllele Technology Enables Objective Analysis of Previously Unusable DNA Evidence
By Dr. Mark W. Perlin, Cybergenetics
Dr. John Yelenic was found murdered in his Blairsville, Pennsylvania home in 2006. His fingernails contained largely his own DNA, but also a small amount of DNA from someone else—possibly deposited when he scratched his assailant in self-defense. Indeed, this minor component of the DNA mixture tied suspect Kevin Foley to the crime, with a match statistic a forensic expert said was 13,000.
DNA mixture data can be hard for human experts to interpret. Their laboratory protocols simplify such data and typically understate the match number. Foley's defense attorney said that the fingernail evidence did not rule out other suspects, since there was a one in 13,000 chance that the DNA came from someone other than his client.
Human expert evaluation of DNA evidence can be challenging, even on simpler samples. The analyst performing the examination requires significant training, and the review process is slow and tedious. Human interpretation methods may not eliminate natural examination bias. Heuristic approaches that truncate data can rob the evidence of much probative value.
Today, most DNA samples are not simple. They can contain little DNA, exhibit degradation, or mix together the DNA of several people. These factors compound the data analysis difficulties. Sometimes expert analysts are unable to draw a conclusion, despite expending considerable effort. As a result, valuable evidence to convict the guilty or exonerate the innocent becomes unusable in court.
Cybergenetics TrueAllele® technology, developed with MATLAB®, uses signal processing and advanced statistical methods to extract identification information from DNA data. TrueAllele's probabilistic approach is more thorough, more objective, and faster than human analysis. These advantages let crime labs extract information from previously inconclusive samples, and reduce backlogs of evidence awaiting review. In the Foley case, TrueAllele enabled a million-fold improvement over the human expert's 13,000 estimate, objectively computing a persuasive 189 billion DNA match statistic that helped secure a conviction.
DNA Identification Glossary
DNA: A linear information molecule that encodes life's operating system and programs. DNA is written in an alphabet of four chemical letters (A, C, G, and T).
Chromosome: A large package of DNA molecules residing in a cell's nucleus. Human DNA comprises 23 chromosome pairs, with one copy inherited from each parent.
Locus: A location on a chromosome that codes for a gene or some other DNA sequence.
Allele: A DNA sentence at a genetic locus. An individual has two alleles (one from the mother and one from the father) at every locus, except on the X and Y sex chromosomes.
Genotype: The genetic composition of a cell or individual. At a particular locus, the genotype of an individual is an allele pair.
Identification: Distinguishing one individual from another by using naturally occurring genotype variation.
Processing Simple and Mixed DNA Samples
When data from a DNA sequencer is plotted, an allele pair is apparent as one or two primary peaks (Figure 1). The peak’s location along the x-axis identifies the allele, while its height along the y-axis reflects the DNA quantity. When the DNA data comes from a single individual, an analyst can easily infer the individual’s genotype from the peaks.
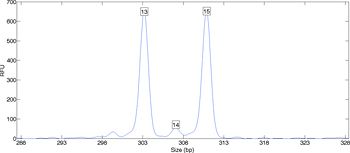
When a sample contains DNA from more than one person, the relationship between data and genotype may be less evident. The laboratory data contains multiple peaks that indicate contributing alleles and their relative amounts (Figure 2). A peak height counts the number of amplified DNA molecules. Such counting data varies between repeated experiments, in accordance with the laws of probability and chemistry.
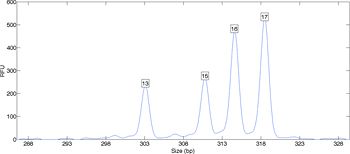
In an attempt to address this data variation, human examination of DNA evidence applies "thresholds." Each laboratory sets its own threshold level, based on an internal calibration. Peaks with a height above this threshold are accorded equal weight, while less use is made of peaks below the threshold. These thresholds do not work well with count data and its variation. Informative DNA samples often end up miscategorized as inconclusive, and are not reported. A more accurate approach is to use computers and probability to mathematically model peak height variance as a parameter of the evidence data.
Using MATLAB to Analyze Complex DNA Samples
TrueAllele technology uses MATLAB, Signal Processing Toolbox™, and Statistics and Machine Learning Toolbox™ to mathematically separate mixed DNA data into individual contributors and their respective genotypes. Since the solution may be uncertain, the inferred genotype values are assigned probabilities.
A DNA sequencer generates laser-detected fluorescent data as a one-dimensional signal, a multicolored multiplex of many loci. A TrueAllele analysis module developed with Signal Processing Toolbox processes the signal data to remove artifacts, classify the peaks, determine peak sizes and heights, and perform other quality checks.
After the initial analysis, TrueAllele interprets the data using a probability model. This model incorporates several hundred variables, including the unknown genotypes of individuals contributing to the sample, DNA quantities, amplification artifacts that distort the signal, and the uncertainties of these variables. Many of the variables are hierarchical, which means they include submodels, each with their own parameters and uncertainties. Starting from the DNA data, TrueAllele solves the model through Markov chain Monte Carlo (MCMC) statistical sampling, using a Metropolis-Hastings algorithm developed with Statistics and Machine Learning Toolbox.
To interpret DNA evidence, TrueAllele proposes 100,000 different combinations of possible values for solution space variables and evaluates how well each proposed solution explains the DNA data. The MATLAB based software then calculates probability densities to produce a probability distribution over the feasible solutions. Solutions that more accurately describe the observed data have higher probability, while poorer explanations have lower probability.
For some samples, the computer can mathematically separate the mixture into virtually single-source components, with a high probability assigned to each genotype. For other samples, the results are less certain and yield genotypes with more diffuse probabilities. Regardless, the genotype answer is a probability distribution, objectively inferred solely from the evidence.
When the data supports a match between the evidence and a suspect’s genotype, the TrueAllele model enables the analyst to calculate a DNA match statistic. To form such a match statistic, or "likelihood ratio," the MATLAB program compares a genotype inferred from the evidence with a reference genotype from the suspect. To eliminate the possibility of examination bias, this comparison is done only after computer genotype inference has completed.
The match calculation includes a third genotype representing the random population, which provides the denominator needed to compare a probability of match with coincidence. Stating the mathematical result in plain language, a forensic scientist can report, for example, that “a match between the evidence item and the suspect is a quadrillion times more probable than coincidence.”
Developing a User Interface and Adding Database Support
To present TrueAllele results in pictures that are intuitive to a scientist, lawyer, or juror, Cybergenetics used MATLAB to develop a Visual User Interface (VUIer™) tool. The VUIer displays visual representations of key variables, such as the data, mixture weights, genotype contributor probabilities, and match strength (Figure 3). VUIer enables a "what if" analysis of alternative genotypes and mixture possibilities that are useful in teaching. The user interface calculates likelihood ratios and confidence intervals, and can generate a DNA match report.
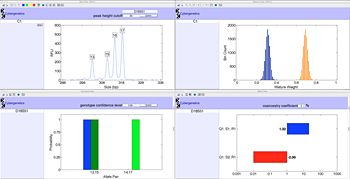
Cybergenetics used MATLAB Compiler™ to package the VUIer client into a standalone executable program. This user interface client is cross-platform and runs on both the Mac OS X and Microsoft® Windows® operating systems. The TrueAllele server performs MCMC genotyping calculations in parallel on multiple computers that run the Linux® operating system. MATLAB enables development in a single environment that can be deployed to three different platforms.
The VUIer client software accesses a TrueAllele database server via Database Toolbox™. This PostgreSQL database serves as a repository for DNA data, interpretation requests, and results. The central server database autonomously coordinates the system's operation through a custom supervisory expert system written in MATLAB.
The TrueAllele system provides DNA database matching capability that can help solve crimes, find missing people, or identify human remains. To solve a cold case, the database system compares genotypes inferred from case evidence with reference genotypes from thousands of potential suspects. The TrueAllele intelligence database is highly sensitive and specific (unlike government-supplied software) since it uses mathematics to represent genotypes and calculate match strength.
Making the World a Safer Place
TrueAllele's reliability has been extensively validated and has withstood court admissibility challenges. The system has been used in the United States and internationally in over a hundred cases, for crimes including rape, homicide, abduction, and terror. TrueAllele was used in the World Trade Center disaster to help identify victim remains. Kevin Foley's unsuccessful appeals of his life sentence for Dr. Yelenic's murder led to Pennsylvania Superior and Supreme Court rulings that have established a statewide TrueAllele precedent.
Whether at Cybergenetics or in crime laboratories, TrueAllele's MATLAB interpretation of previously unusable biological evidence can now compute accurate DNA identification information.
Published 2013 - 92093v00